治験実施の流れ
1.治験実施のための手続き

2.治験実施のための手続き
IRBで「治験の実施」が患者さんの人権や安全の保護及び科学性等に問題がないかどうかの診査をします。
3.治験の実施に関する研究の契約
IRBで承認し、施設長が了承したら、製薬企業と「治験の実施」に関する契約を結びます。
4.スタートアップ・ミーティング
治験責任医師・分担医師(以下、医師)と治験コーディネーター(CRC)、関連部署、製薬企業の間でミーティングを重ね、治験が円滑に進むよう話し合います。
5.治験の開始
医師やCRCが、説明文書により、患者の皆様に治験の説明をし、同意されたら治験を開始します。
6.直接閲覧(モニタリング)の実施
治験期間中でも必要に応じて、製薬企業は医療機関で治験が正しく行われているかを調べます。
7.症例報告書の作成・提出
症例報告書を作成し製薬企業に提出します。
8.直接閲覧(モニタリング)・監査の実施
製薬企業は、医療機関で治験が正しくおこなわれていたか、医師から提出された症例報告書に記入漏れがないかを調べます。
9.治験の終了
治験責任医師は治験終了報告書をIRBへ提出し報告します。
治験のルール(GCP)
厚生労働省が定める「医薬品の臨床試験の実施の基準」(Good Clinical Practice)
治験実施にあたり、治験に参加する人の人権や安全性、プライバシーを守るために厳しいルールが定められています。
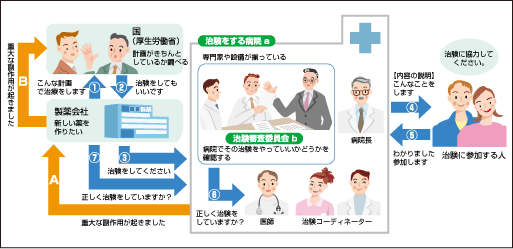
国(厚生労働省)への治験内容の届出(①)
製薬会社は、治験の内容が記載された「治験実施計画書」を国(厚生労働省)に届け出ます。「治験実施計画書」は製薬会社と専門家が協議して作成されます。
国(厚生労働省)での調査(②)
届出を受けた「治験実施計画書」の内容に問題がないか(人権と安全性に配慮されているかなど)を調査し、問題があれば修正を指導します。
製薬会社が病院に治験を依頼(③)
インフォームド・コンセント(④⑤)
治験の目的やこれまでに分かっている「くすりの候補」の効き目、副作用などが記載された同意説明文書をもとに、治験担当医師などが、治験参加を希望する人に説明を行います。文書により同意した人のみが治験に参加します。(インフォームドコンセント)
治験の適正な実施の確認(⑥⑦)
「治験実施計画書」や治験のルール(GCP)を守って、治験が適正に行われているかを確認します。
専門家や設備が揃った病院(a)
治験を行う病院は、専門の医師をはじめとするCRC(治験コーディネーター)などのスタッフが揃っていて、十分な設備が整ってなければなりません。
治験審査委員会での確認(b)
病院では、治験に参加する人の人権や安全性を守って「くすりの候補」の治療効果を科学的に調べることができるかなどを、慎重に治験審査委員会で確認をします。この治験審査委員会とは、医師などの医療関係者だけではなく、病院と利害関係がない人と専門外の人も必ず参加します。
重大な副作用の国(厚生労働省)への報告(AB)
治験中に起きた重大な副作用は、病院から製薬会社に連絡され、さらに製薬会社から国(厚生労働省)に報告されます。治験に参加している人の安全性を守るため、必要な場合には治験の見直しが行われます。
社団法人 日本医師会 治験促進センターパンフレットより
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()